

What is Pharmacokinetics?
The term Pharmacokinetics is derived from two words: pharmacon means drugs and kinetics means movement of drugs.
The simple meaning of pharmacokinetics is “what the body does to the drugs”. It deals with the study of the movement of drugs in, through, and out of the body.
The pharmacokinetics include the following process ADME i.e. absorption(A), Distribution (D), Metabolism (M), and Excretion (E).
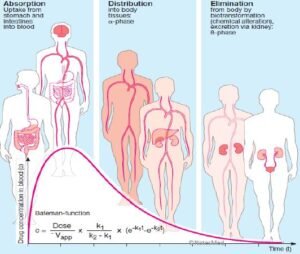
Absorption (@ Pharmacokinetics)
The movement of drugs from the administration site into the systemic circulation (bloodstream) in the body is called absorption.
Factor affecting drugs absorption
- Physicochemical properties of drugs
- Physical state: liquid form >solid formulation
- Lipid -soluble and unionized>water-soluble and ionized form.
- Particle size: smaller size> larger size, for example, digoxin, griseofulvin, etc are well absorbed from the gut and
- produce better effects But some anthelmintics drugs have larger particle size when they are poorly absorbed in the GI tract hence produce a better effect.
- Disintegration time:
It is defined as the time required for the formulation to break down into small particles and its variation may affect the bioavailability.
- Dissolution time:
It is defined as the time is required for the particles to go into solution and the shorter the time and better is the absorption. - Formulation:
- Route of drugs administration
The intravenous route bypasses the process of absorption as it directly enters the systemic circulation. - pH and ionization:
The strongly acidic (heparin) and basic (aminoglycosides) drugs generally ionized at all pH, hence they are poorly absorbed in your body. - Food:
Food present in your stomach can influence (affect) the absorption of some drugs. Food reduced the absorption of drugs such as rifampicin, levodopa, etc hence, they are taken on an empty stomach for better effects.
Fatty meals increase the absorption of griseofulvin and milk & milk products which decrease the absorption of tetracyclines.
- Presence of other drugs:
Two or more drugs concurrently administered may affect their absorption, for example, ascorbic acid increases the absorption of oral iron and antacids decrease the absorption of tetracyclines.
- Area of the absorbing surface:
In normally better-absorbed drugs in the small intestine due to the fact that of a larger surface area. When the gut is resected that leads to decreased absorption of drugs due to decreased surface area in the gut. - Gastrointestinal and other diseases
Bioavailability
It is defined as the fraction of a drug that reaches systemic circulation (bloodstream) from a given dose in unchanged form.
When we administer a drug orally, it is first absorbed into the portal circulation & reaches the liver. Here, some of the drugs may be metabolized (first-pass metabolism or pre-systemic metabolism) and the rest of the drug reaches the systemic circulation.
Thus absorption and first-pass metabolism are 2 important determinants of bioavailability.
The Intravenous (IV) route of drug administration gives 100% bioavailability because it directly enters the systemic circulation. This term is commonly used for drugs given by oral route.
First-pass metabolism (First-pass effect, pre-systemic elimination)
When drugs are administered orally, they have to pass via the gut wall and enter into the portal vein and reach the liver and reach the systemic circulation.
During this passage, some drugs get metabolized and are removed or inactivated before they reach the systemic circulation which is called the first-pass metabolism.
Consequences of high first-pass metabolism
- Drugs that undergo extensive first-pass metabolism are administered parenterally, for example, lignocaine is administered IV in ventricular arrhythmias.
- The dose of a drug required for the oral route of drug administered is more than that given by other systemic routes, for example, nitroglycerin.
Bioequivalence
If two drugs have the same formulations that produce equal bioavailability, they are called bioequivalent.
Distribution (@ Pharmacokinetics)
It is defined as the reversible transfer of drugs between fluids compartments in the body. After drug absorption, a drug enters the systemic blood circulation & is distributed in the body fluids.
The apparent volume of distribution (aVd)
It is described as the hypothetical volume of body fluid into which a drug is uniformly distributed at a concentration that is equal to that in plasma, assuming the body to be a single compartment.

If more drugs are entering the tissues, it has a higher volume of distribution in the body and vice-a-versa. It also depends on several factors such as lipid solubility (high volume of distribution) and plasma protein binding(reduced volume of distribution).
The volume of distribution (Vd)
It is defined as the ratio of the dose administered (IV) to the plasma concentration of a drug. When the volume of distribution (Vd) is more, that means more amount of drug is in the tissues and less is in the plasma in the body.
Metabolism (biotransformation) (@ Pharmacokinetics)
It is defined as the chemical alteration of the drug in a living organism and the primary site of drug metabolism in the liver and other sites are the GI tract, kidney, lungs, skin, placenta, and blood.
During metabolism, the drug normally converts lipid-soluble and unionized compounds into water-soluble and ionized compounds in the body.
- Active drug to inactive metabolites, for example, phenobarbitone to hydroxy phenobarbitone
- Active drug to active metabolites, for example, codeine into morphine, etc.
- Inactive drug (prodrug) to active metabolites, for example, levodopa to dopamine, etc.
Prodrug
When an inactive form of the drug is converted into the active form after metabolism.
Uses of prodrugs
- Improve bioavailability of drugs
- Duration of action prolongs
- Improve the taste of drugs
- It provides site-specific drug delivery
Pathway of drug metabolism
There are two phases of drug metabolism, they are phase I (Non-synthetic reactions) and phase II (synthetic reactions).
Phase I reactions:
This non-synthetic reaction includes oxidation, reduction, hydrolysis, cyclization, and decyclization in the body. At the end of the phase-I reaction, the metabolites may be in an inactive form or inactive form.
Phase II reactions:
It is a phase of conjugation reactions. The phase II reactions include glucuronidation, acetylation, sulphation, methylation, glutathione conjugation, and glycine conjugation.
If the phase-I metabolites are polar in nature, then it is excreted in urine or bile. However, most metabolites are lipophilic and undergo conjugation reactions.
Factors affecting metabolism
- Age
- Diet
- Diseases
- Genetic factors (pharmacogenetics)
- Stimulation of drugs administration including enzyme induction or inhibition
Enzyme induction
The repeated administration of some drugs increases the synthesis of microsomal enzymes; this is called enzyme induction.
Enzyme inducer drugs are @GPRS Cell Phone means
- G-Griseofulvin
- P-Phenytoin
- R-Rifampin
- S-Smoking
- Cell- Carbamazepine
- Phone-Phenobarbitone
Enzyme inhibition
Some drugs inhibit the activity of drug-metabolizing enzymes and are known as enzyme inhibitors. The enzyme inhibition is a rapid process as compared with enzyme induction. Enzyme inhibitors drugs include
@Vitamin K Cannot Cause Enzyme Inhibition
- Vitamin – Valproate
- K – Ketoconazole
- Cannot – Cimetidine
- Cause – Ciprofloxacin
- Enzyme – Erythromycin
- Inhibition – INH
Excretion (@ Pharmacokinetics)
The major site of excretion is the kidney and other organs include lungs, feces, bile, skin, saliva, milk, etc. The excretion through kidneys occurs by glomerular filtration, tubular reabsorption, and tubular secretion in the body.
Glomerular filtration
It depends on the plasma protein binding and renal blood flow. Drugs with small molecular sizes are more readily filtered.
The extent of filtration is directly proportional to the glomerular filtration rate and to the fraction of unbound drugs in the plasma of the body.
Passive tubular reabsorption
It depends on lipid solubility. If a drug is highly lipid-soluble, more of it will be reabsorbed and less will be excreted in the body and vice-versa.
As lipid solubility depends on its ionization, the ionized drug will be excreted by the kidney rapidly.
Active tubular secretion
It is not dependent on lipid solubility or plasma protein binding. It is a carrier-mediated active transport that needs energy. Drugs having similar physicochemical properties due to the same carrier system (relatively nonselective)
Pharmacokinetics parameter
Includes following parameter
- Bioavailability
- Volume of ditribution
- Plasma half-life (t1/2)
- Clearance
Plasma half-life
It is defined as the time taken for the plasma concentration of the drug to decreased by about 50% of its original value. For example, the plasma half-life for aspirin is four hours.
Significance of plasma half-life
- It helps to determine the duration of action of drugs
- It helps to determine the frequency of drug administration
- It helps to estimate the time required to reach the steady-state
Clearance
It is defined as that volume of plasma from which the drug is removed in unit time.

First-order kinetics
A constant fraction of the drug in the body is eliminated (excreted) per unit time period. The rate of drug elimination is directly proportional to its plasma concentration and the plasma half-life of the drugs is always remains constant.
The drug will be almost completely eliminated in four to five plasma half-lives if administered at a constant rate at each half-life. Most of the drugs are follow first-order kinetics.
Zero-order kinetics
A constant amount of drugs in the body eliminated (excreted) per unit time period. The rate of elimination is independent of plasma drug concentration, for example, ethanol is eliminated from the body at the rate of about 10mL/hr.
The plasma half-life (t1/2) of the drug following zero-order kinetics is never constant.
Steady-state concentration
If a constant dose of a drug is given at a constant interval at its plasm half-life (t1/2), the plasma concentration of the drug increases due to its absorption and falls due to elimination or excretion in each interval.
The final amount of drugs eliminated will be equal to the number of drugs administered in the dosing interval. It is generally attained after approximately 4 to 5 half-lives.
Download Pharmacokinetics pdf file



